We are excited to be part of the 4thGlobal Precision Medicine & Biomarkers
Leaders Summit [http://www.global-engage.com/event/precision/]taking place on
September 21-22, 2017 in Munich, Germany.
Data4Cure will co-host an immunooncology roundtable with Dr. Pallavi Sachdev,
Director, Oncology Biomarker Research, Clinical Research, Eisai.
Please join us




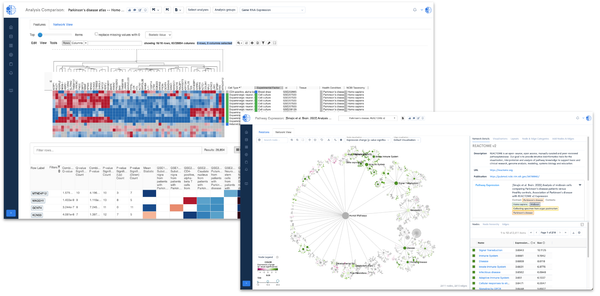





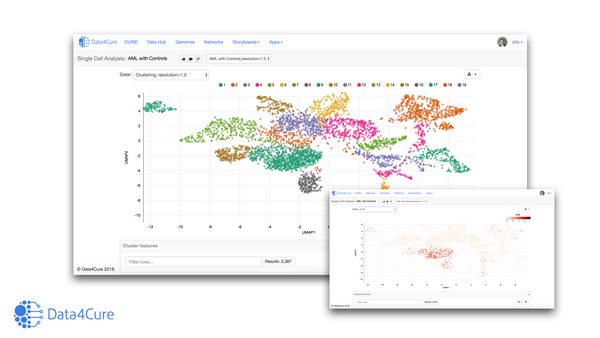
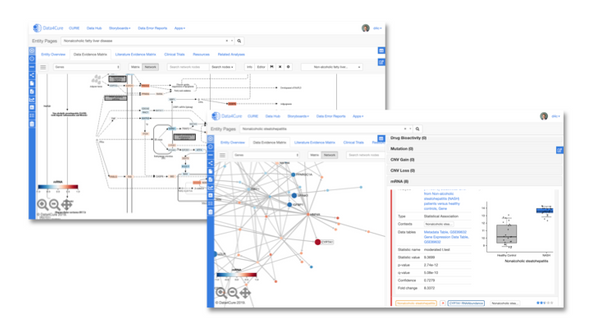
![Mapping immunotherapy resistance pathways with biomedically-informed AI [Data4Cure @ PMWC 2019]](/content/images/size/w600/wordpress/2019/03/JD-PMWC-e1552187919456.png)